Alopecia areata (AA) is a common, chronic autoimmune disorder characterized by non-scarring hair loss that significantly affects approximately 2% of the global population. The disease imposes a substantial psychosocial burden, often linked to anxiety and depression. Pathogenically, AA is driven by the collapse of hair follicle immune privilege and the activation of autoreactive CD8+ NKG2D+ T lymphocytes, leading to the release of pro-inflammatory cytokines like interferon-γ (IFN-γ) that perpetuate follicular damage. While immunological and genetic factors play roles, the gut microbiota has emerged as a key regulator of systemic immunity and a potential contributor to autoimmunity. Gut microbial changes can modulate cutaneous inflammation via the gut–skin axis, either through microbial metabolites entering systemic circulation or by activating mucosal immune responses with peripheral effects. Given that dysbiosis—a disruption in the normal composition of the microbiota—has been associated with various autoimmune conditions, investigating intestinal dysbiosis in AA was considered highly plausible. Furthermore, microbiome profiling combined with artificial intelligence (AI)-based classification models offers a promising non-invasive source for identifying disease-specific diagnostic and prognostic biomarkers.
Methods
The study analyzed fecal samples from patients with AA and healthy controls using 16S rRNA sequencing to characterize gut microbiota composition. Data were processed using QIIME2 and MicrobiomeAnalyst platforms to assess microbial diversity, differential abundance, and co-occurrence networks. To develop a non-invasive diagnostic tool, supervised machine learning models, specifically Random Forest, were trained and validated using 11 selected genera as features. The Synthetic Minority Over-sampling Technique (SMOTE) was applied to address the dataset’s imbalance (102 healthy samples vs. 19 AA samples) during the model training phase.
Section-Wise Key Points
Microbial Community Analysis and Diversity
• Beta diversity analysis, measured using the Bray–Curtis distance, revealed significant differences in the microbial community structures between AA patients and healthy controls (FDR-corrected p-value of 0.001).
• In contrast, alpha diversity indices, including the Chao1 and Shannon indices, showed no statistically significant differences between the two groups.
Differential Abundance and Dysbiotic Signatures
• AA patients showed a significant depletion of immunoregulatory commensals, notably the beneficial genera Faecalibacterium and the Eubacterium eligens group, which are known producers of anti-inflammatory short-chain fatty acids (SCFAs).
• Conversely, AA patients exhibited an enrichment of several pro-inflammatory genera, including Methanobrevibacter, Collinsella, and Ruminococcus gnavus.
• Specific bacterial families showing increased abundance in AA patients included Methanobacteriaceae, Coriobacteriaceae, Peptostreptococcaceae, and Eggerthellaceae.
Microbial Network Correlations
• Healthy controls showed sparse and stable correlation networks, typically consisting only of positive associations between commensals.
• AA patients displayed a significantly increased number and complexity of microbial correlations.
• Key hub taxa involved in regulating the complex networks in AA patients included Methanobrevibacter, Christensenellaceae R-7 group, and Eubacterium ventriosum group, suggesting ecological instability in response to inflammation.
Machine Learning (AI) Modeling for Diagnosis
• A supervised machine learning Random Forest model, utilizing information from 11 microbial genera, was highly effective at classifying AA patients from healthy controls.
• The model achieved a 92% accuracy in distinguishing AA from controls during validation, underscoring the strong association between gut microbiome composition and the disease.
• The most influential taxa identified for classification were Collinsella and Methanobrevibacter.
This study offers the first comprehensive characterization of gut microbiome alterations in AA that integrates 16S rRNA sequencing with microbial network inference and predictive machine learning modeling. The high predictive performance of the Random Forest model (92% accuracy) highlights the gut microbiome’s potential as a robust diagnostic biomarker. The research successfully quantified diagnostic performance using cross-validated metrics and identified biologically plausible microbial features, providing testable mechanistic hypotheses regarding AA pathogenesis.
The findings support the relevance of the gut–immune–skin axis in AA pathogenesis and have significant translational implications. First, the microbial signatures identified could form the basis of an objective, non-invasive diagnostic aid for the early identification of alopecia areata. Pending external validation, these microbiome-based classifiers could potentially assist clinicians in early disease detection and support clinical stratification by disease severity. Second, the identified dysbiosis—characterized by the loss of SCFA producers and the expansion of pro-inflammatory taxa like Collinsella and Ruminococcus gnavus—suggests potential avenues for future therapeutic interventions. Microbiome-targeted interventions, such as dietary modulation, probiotics, prebiotics, or fecal microbiota transplantation (FMT), could be explored as adjunctive therapies in controlled clinical trials. Further research must employ longitudinal studies and multiomic approaches, such as metagenomics and metabolomics, to confirm generalizability, establish causality, and fully define the clinical utility of these findings.
The findings suggest that the gut microbiome may act as a crucial amplifier of AA immune responses, consistent with the immune profile of AA which involves CD8+ and Th17 pathways. Understanding these microbial shifts is like reading the barometer of the body’s internal immune climate, providing a predictive measure for a disease whose primary drivers manifest externally on the skin.
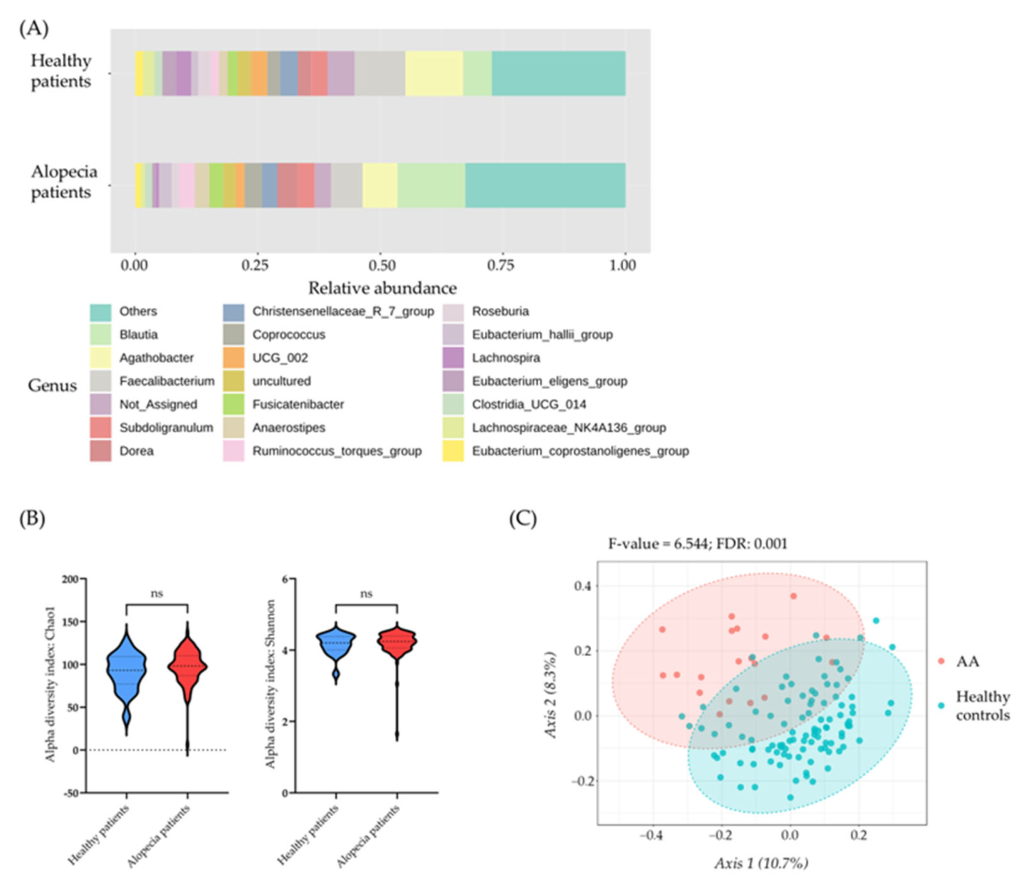
Microbial community profile in healthy individuals and patients with alopecia: (A) Bar plot showing the microbiome composition at the genus level (percentage of abundance) in healthy individuals and patients with alopecia. (B) Alpha diversity measured using the Chao1 index (left) and Shannon index (right) for each sample within the study groups: healthy individuals and patients with alopecia. Statistical differences were assessed using the value of each index per sample and analyzed by One-Way ANOVA, followed by Tukey’s test for multiple hypothesis correction. (C) Representation of beta diversity for each microbial community using the Bray–Curtis index. Statistically significant differences in beta diversity among the different pathologies were assessed by pairwise comparisons using the PERMANOVA algorithm. Differences were considered statistically significant at p < 0.05 (p > 0.05 = ns).